[[{“value”:”

Designing accurate all-atom protein structures is a critical challenge in bioengineering, as it involves generating both 3D structural data and 1D sequence information to define side-chain atom placements. Most approaches currently rely heavily on resolved experimentally determined structural datasets, which are scarce and biased, thereby limiting exploration of the natural protein space. Moreover, these approaches typically compartmentalize the processes of sequence and structure generation, which results in inefficiencies and an insufficient integration of multimodal aspects. Addressing these obstacles is crucial for progressing protein design, and facilitating advancements in drug discovery, molecular engineering, and synthetic biology.
Conventional methods of designing proteins rely on backbone-structure prediction or inverse-folding models to infer sequences from known structures. These methodologies are limited by the separation of modalities, given that numerous techniques address sequence and structure as distinct tasks, thereby complicating their integration. A significant dependence on experimentally determined structural datasets precludes a considerable number of non-crystallizable proteins and restricts generalizability. The process of alternating between structure prediction and sequence generation prolongs runtime and heightens the risk of error propagation. Current tools also face challenges in being able to fit large-scale, function-specific protein designs due to rigid structures and limitations in the datasets. These limitations limit the scope of existing methodologies, particularly regarding the design of proteins that require high multimodal fidelity and variation.
Researchers from UC Berkeley, Genentech, Microsoft Research, and New York University introduce PLAID (Protein Latent Induced Diffusion), a generative framework that synthesizes sequence and all-atom protein structures by leveraging latent diffusion. Such a framework uses ESMFold-derived cohesive latent space for smooth multimodal integration and reduces dependence on outside predictive models. This approach increases the data distribution by 2–4 orders of magnitude over structural datasets to cover natural protein diversity better. It applies a Diffusion Transformer architecture designed to be hardware-aware and scale up for conditional multimodal protein generation. Its function and organism-conditioned samples attain high structural fidelity and biophysical relevance across a wide spectrum of protein design needs. These features describe massive strides forward toward making diverse, scalable, and application-driven protein generation possible.
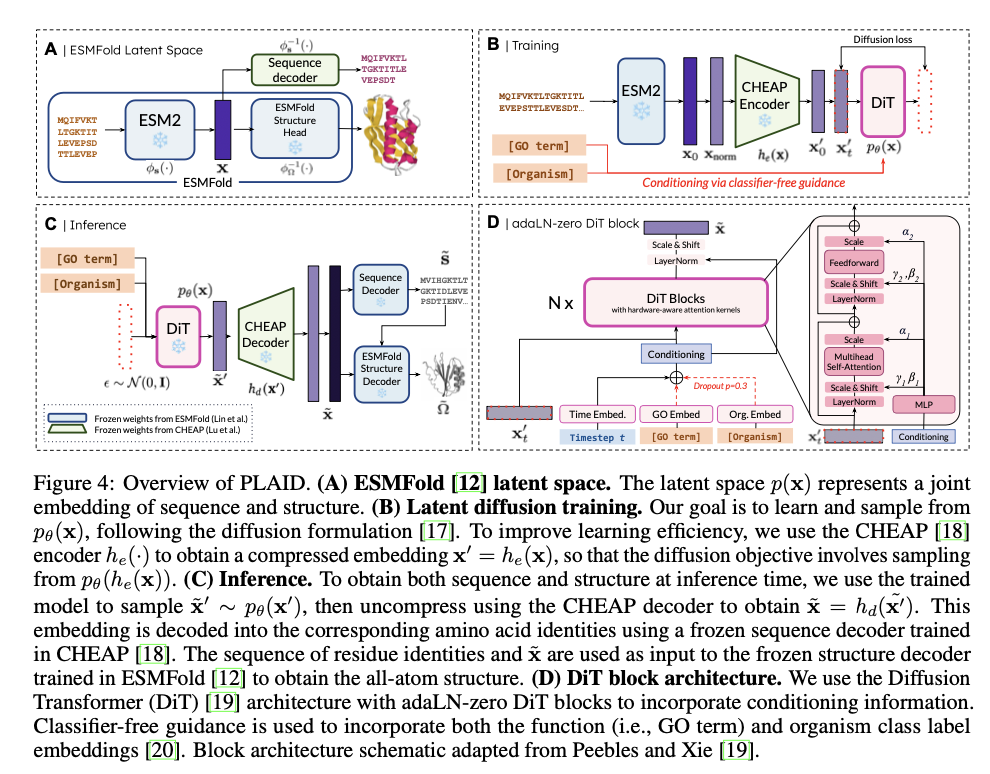
PLAID operates by correlating protein sequences to a reduced latent space, fine-tuned for efficient diffusion and generation. Indirect acquisition of structural constraints occurs with the use of pre-trained ESMFold weights, owing to abundant annotated sequence data in databases like Pfam. A discrete-time diffusion framework systematically enhances latent embeddings while integrating classifier-free guidance for conditioning related to function and organism. It incorporates a CHEAP autoencoder for dimensionality reduction and Diffusion Transformer blocks for denoising, thus making it scalable for long protein sequences. Metrics include cross-modal consistency, self-consistency, and conformity to natural protein biophysical distributions, validating the quality and biological relevance of generated samples.
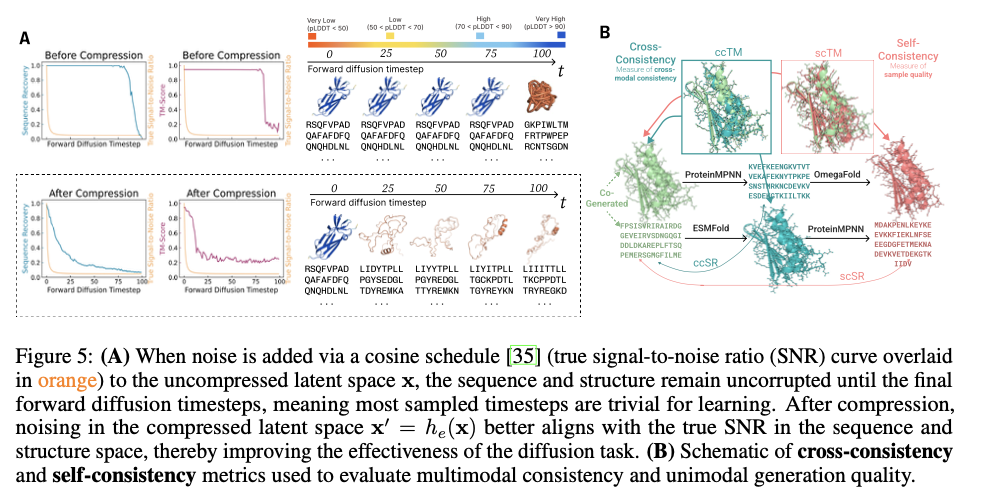
This innovative framework demonstrates substantial advancements in generating biologically relevant and structurally accurate protein designs. PLAID is highly cross-modal consistent and therefore demonstrates substantial improvements in the alignment of generated sequences and structures and also presents an improved designability relative to previous approaches. The proteins produced demonstrate robust structural fidelity, capturing the biophysical properties such as hydropathy, molecular weight, and diversity in secondary structure, thus closely resembling the natural proteins. Also, function-conditioned samples demonstrate elevated specificity, precisely reproducing motifs of active sites and patterns of hydrophobicity, and with preserved sequence diversity. In terms of quality, diversity, and novelty metrics, PLAID demonstrates a consistent superiority over baseline models, yielding the greatest quantity of distinct and designable protein clusters that exhibit realistic structural and functional characteristics.
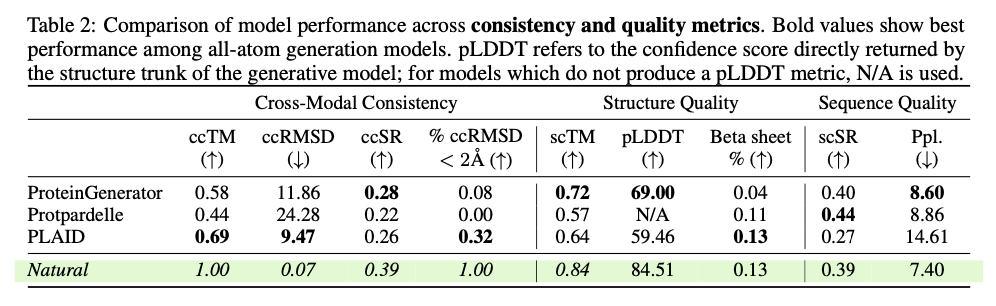
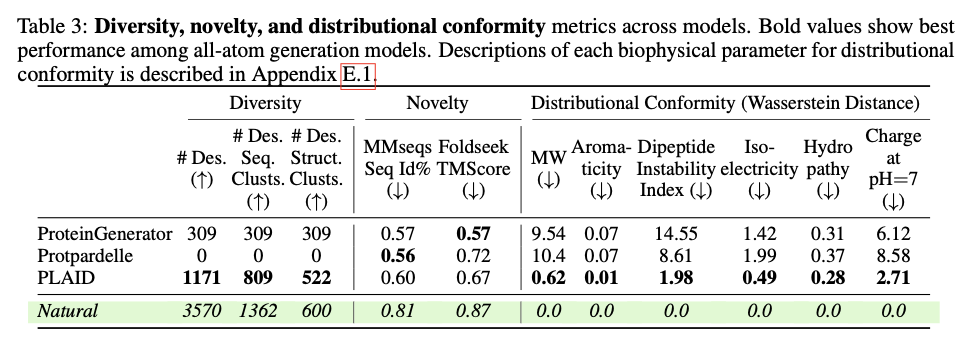
PLAID presents a revolutionary method for protein design by focusing on scalability, data availability, and multi-modality integration of major challenges. This way, it manages to link sequence and structure generation by applying sequence-based training and latent diffusion for creating diverse, accurate, and biologically relevant protein designs. This approach can potentially be used to further applications in molecular engineering, therapeutic development, and synthetic biology and open up the doors for new solutions for complex biological problems.
Check out the Paper and GitHub Page. All credit for this research goes to the researchers of this project. Also, don’t forget to follow us on Twitter and join our Telegram Channel and LinkedIn Group. Don’t Forget to join our 60k+ ML SubReddit.
 Trending: LG AI Research Releases EXAONE 3.5: Three Open-Source Bilingual Frontier AI-level Models Delivering Unmatched Instruction Following and Long Context Understanding for Global Leadership in Generative AI Excellence….
Trending: LG AI Research Releases EXAONE 3.5: Three Open-Source Bilingual Frontier AI-level Models Delivering Unmatched Instruction Following and Long Context Understanding for Global Leadership in Generative AI Excellence….The post PLAID: A New AI Approach for Co-Generating Sequence and All-Atom Protein Structures by Sampling from the Latent Space of ESMFold appeared first on MarkTechPost.
“}]] [[{“value”:”Designing accurate all-atom protein structures is a critical challenge in bioengineering, as it involves generating both 3D structural data and 1D sequence information to define side-chain atom placements. Most approaches currently rely heavily on resolved experimentally determined structural datasets, which are scarce and biased, thereby limiting exploration of the natural protein space. Moreover, these approaches
The post PLAID: A New AI Approach for Co-Generating Sequence and All-Atom Protein Structures by Sampling from the Latent Space of ESMFold appeared first on MarkTechPost.”}]] Read More AI Paper Summary, AI Shorts, Applications, Artificial Intelligence, Editors Pick, Machine Learning, Staff, Tech News, Technology